|
|


|
|
|
|



A. Progress Statement
As stated in the proposal, current
computation of biomolecular solvation faces many fundamental
limitations and sever challenges, such as ad hoc assumptions about
solvent-solute interfaces to define some of the most important components of
the solvation model, and about linear and local solvent response to all solute
perturbation. Our goal is to develop geometric flow (i.e., differential geometry)
approaches to overcome the abovementioned difficulties in implicit solvent
theory and explore the application of the new solvation model. During the last
three years, we have investigated novel geometric
flow approaches for the solvation analysis of small compounds and biomolecules.
We have also developed differential geometry based multiscale
models. Applications are considered to viral surface analysis, charge transport
in ion channels, proton transport, molecular surface
generation. Computational algorithm design and software development are in
progress.
B. Studies and Results
[Differential geometry based solvation models]
We have constructed a novel differential geometry
based solvation model that starts with a total free energy functional [1, 2]:
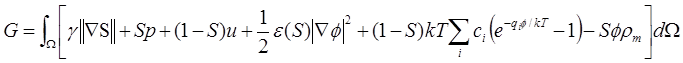
where first three terms are surface energy, mechanical work
and solvent-solute interactions, respectively. The rest terms are the
electrostatic energy. Here, ![]() is the surface
tension, S is a surface
characteristic function, p is the
pressure,
is the surface
tension, S is a surface
characteristic function, p is the
pressure, ![]() is the
dielectric function,
is the
dielectric function, ![]() is the
electrostatic potential, k is the
Boltzmann constant and T is the
temperature,
is the
electrostatic potential, k is the
Boltzmann constant and T is the
temperature, ![]() is the bulk
concentration of ith
ion, and
is the bulk
concentration of ith
ion, and ![]() is charge
source function of the solute. Coupled generalized Poisson-Boltzmann and
geometric flow equations,
is charge
source function of the solute. Coupled generalized Poisson-Boltzmann and
geometric flow equations,
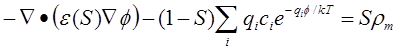
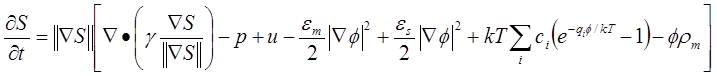
are derived via the variation of the total free energy
functional. We would like to quote two anonymous referees’ comments of our
manuscript [1] as a summary of this work:
Reviewer #1: The authors present a method for simultaneously computing the
electrostatic and 'surface' free energy of a molecule. The approach combines a
finite difference Poisson-Boltzmann treatment of the electrostatics with a
computational geometry treatment of the molecule: In the latter, the surface
bounding the low dielectric interior from the high dielectric solvent is
treated through a 3-D functional S, which takes on a value of 1 or 0 in the two
regions, and smooth varies between these values in the interface. Then van der
Waals, PV and surface free energy terms are introduced as functions of S. The
total energy can then be optimized by simultaneous solution of the PB equation
and the S dependent energy terms through variation of the phi (potential) and S
functionals. This provides a self consistent treatment of the dielectric
boundary in the PB and the surface free energy terms.
This is a novel and important advance in the continuum treatment of
molecular free energies. The ms. explains the basic ideas clearly and
thoroughly, and there is a thorough and systematic exploration of the best way
to solve the coupled surface-electrostatic equations numerically. The results
are compared to appropriate previous calculations, and the results appear to be
very good. This is an excellent piece of work.
I only had a few small comments
[break].
Fig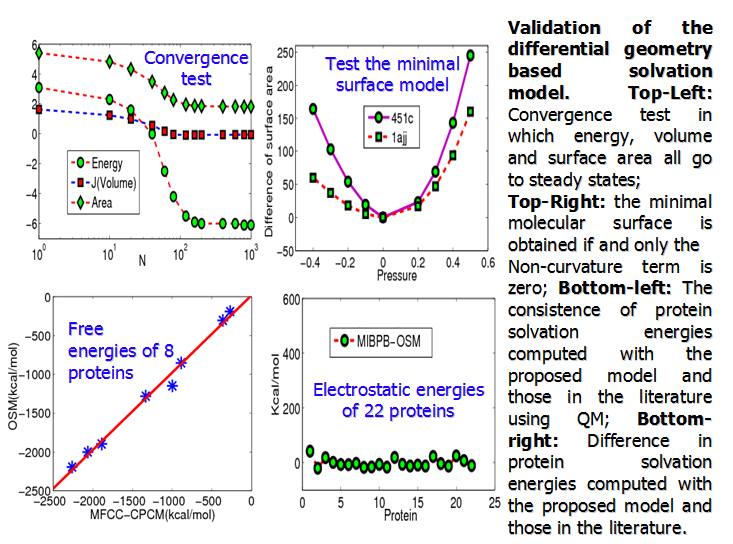
Figure 1. Differential geometry based solvation model [1,2].
Reviewer #2: Synopsis: A novel generalized
Poisson-Boltzmann equation is derived, based on total free energy functional that couples polar and non-polar contributions. The
formalism also yields a generalized geometric flow equation for construction of
realistic solvent-solute boundaries. The new model is tested against
experimental solvation free energies of several small molecules, and against QM
energies for 22 proteins. Accuracy,
stability and robustness issues are thoroughly discussed.
Over-all: this is a very interesting, thorough work. Given the importance
of the implicit solvent framework for molecular modeling, and the pressing need
for improving its accuracy, this study is very relevant.
Recommendation: Publish with minor (non-mandatory) revisions.
This work has already covered a lot of ground and addressed most of key issues that one would expect in a paper describing a novel method. Addressing additional issues listed below will, in my opinion, strengthen the paper, but should not be considered as mandatory [break].

Figure 2. Surface electrostatic potential
[11].
Atomic partial charges are used in our earlier
differential geometry based solvation models, which depends on existing
molecular mechanical force field software packages for partial charge
assignments. As most force field models are parameterized for certain class of
molecules or materials, the use of partial charges limits the accuracy and
applicability of our earlier models. Moreover, the fixed point charge does not
account for the charge rearrangement during the solvation process. The present
work proposes a differential geometry based multiscale
solvation model which makes use of electron density computed directly from the
quantum mechanical principle. We
construct a new multiscale total energy functional
which consists of not only polar and nonpolar solvation contributions, but also
the electronic kinetic and potential energies. By using the Euler-Lagrange
variation, we derive a system of three coupled governing equations, i.e., the
generalized Poisson-Boltzmann equation for electrostatic potential, the
generalized Laplace-Beltrami equation for solvent-solute boundary, and the
Kohn-Sham equation for electronic structure. We develop an appropriate
iterative procedure to solve three coupled equations and to minimize the
solvation free energy. The present multiscale model
is numerically validated for its stability, consistency and accuracy.
Applications are considered to a few sets of molecules, including a case which
is difficult for existing solvation models. Comparison is made with many other
classic and quantum models. By using
experimental data, we show that the present quantum formulation of our
differential geometry based multiscale solvation
model improves the prediction of our earlier models, and outperforms some
explicit solvation analysis. This work is presented in Ref. [11]
[Differential geometry based
nonpolar solvation models]
Nonpolar
solvation analysis offers a unique setting to test the validity of
solvent-solute interface in the differential geometry based solvation model due
to the absence of the interference of electrostatic effects. Many implicit models have been developed for
nonpolar solvation analysis. However, their
performance is not satisfactory as shown Figure 3. These models rely on
unphysical definitions of solvent-solute boundaries. Based on the differential
geometry, the present work defines the solvent-solute boundary via the
variation of the nonpolar solvation free energy. The solvation free energy
functional of the system is constructed based on a continuum description of the
solvent and the discrete description of the solute, which are dynamically
coupled by the solvent-solute boundaries via dispersion interactions. The first
variation of the energy functional gives rise to the governing Laplace-Beltrami
equation. The present model predictions of the nonpolar solvation energies are
in an excellent agreement with
experimental data, which sheds light on the nature of solvent-solute
boundaries. This work is presented in Ref. [22].

Figure 3. Differential geometry based nonpolar solvation model [22] gives rise to
superb prediction of experimental results.
[Differential geometry based multiscale models]
We have developed a differential geometry based multiscale paradigm to model complex macromolecular systems,
and to put macroscopic and microscopic descriptions on an equal footing [3,21]. Large chemical and biological systems such as fuel
cells, ion channels, molecular motors, and viruses are of great importance to
the scientific community and public health. Typically, these complex systems in
conjunction with their aquatic environment pose a fabulous challenge to
theoretical description, simulation, and prediction. In our approach, the
differential geometry theory of surfaces and geometric measure theory are
employed as a natural means to couple the macroscopic continuum mechanical
description of the aquatic environment with the microscopic discrete atomistic
description of the macromolecule. Multiscale free
energy functionals, or multiscale action functionals are constructed as a unified framework to
derive the governing equations for the dynamics of different scales and
different descriptions. Two types of aqueous macromolecular complexes, ones
that are near equilibrium and others that are far from equilibrium, are
considered in our formulations. We show that generalized Navier–Stokes
equations for the fluid dynamics, generalized Poisson equations or generalized
Poisson–Boltzmann equations for electrostatic interactions, and Newton’s
equation for the molecular dynamics can be derived by the least action
principle. These equations are coupled through the continuum-discrete interface
whose dynamics is governed by potential driven geometric flows. Comparison is
given to classical descriptions of the fluid and electrostatic interactions
without geometric flow basedmicro-macro interfaces.
The detailed balance of forces is emphasized in the present work. We further
extend the proposed multiscale paradigm to
micro-macro analysis of electrohydrodynamics,
electrophoresis, fuel cells, and ion channels. We derive generalized
Poisson–Nernst–Planck equations that are coupled to generalized Navier–Stokes equations for fluid dynamics, Newton’s
equation for molecular dynamics, and potential and surface driving geometric
flows for the micro-macro interface. For excessively large aqueous
macromolecular complexes in chemistry and biology, we further develop
differential geometry based multiscale
fluid-electro-elastic models to replace the expensive molecular dynamics
description with an alternative elasticity formulation. This work is published
in Ref [3] (61 pages) and [21] (55 pages).
[Application to viral surface
analysis]
We have developed a differential geometry-based multiscale
paradigm to model complex biomolecule systems [4]. Viruses are infectious
agents that can cause epidemics and pandemics. The understanding of virus
formation, evolution, stability, and interaction with host cells is of great
importance to the scientific community and public health. Typically, a virus
complex in association with its aquatic environment poses a fabulous challenge
to theoretical description and prediction. In our approach, the differential geometry theory of surfaces and geometric
measure theory are employed as a natural means to couple the macroscopic
continuum domain of the fluid mechanical description of the aquatic environment
from the microscopic discrete domain of the atomistic description of the
biomolecule. A multiscale action functional is
constructed as a unified framework to derive the governing equations for the
dynamics of different scales. We show that the
classical Navier-Stokes equation for the fluid
dynamics and Newton’s equation for the molecular dynamics can be derived from
the least action principle. These equations are coupled through the
continuum-discrete interface whose dynamics is governed by potential driven
geometric flows.
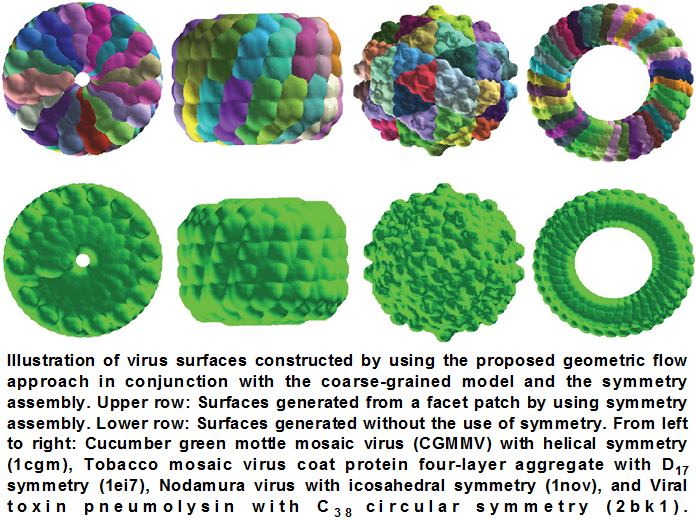
Figure 4. Virus
surfaces [4].
[Application to ion transport
in ion channels]
The Poisson Nernst-Planck (PNP) model is based on a
mean-field approximation of ion interactions and continuum descriptions of
concentration and electrostatic potential. It provides qualitative explanation
and increasingly quantitative predictions of experimental measurements for the
ion transport problems in many areas such as semiconductor devices, nanofluidic systems and biological systems, despite of many
limitations. However, in the PNP model, the number of equations to be solved
and the number of diffusion coefficient profiles to be determined for the
calculation directly depend on the number of ion species in the system, since
each ion species corresponds to one Nernst-Planck equation and one
position-dependent diffusion coefficient profile. In a complex system with
multiple ion species, the PNP can be computationally expensive and parameter
demanding, as experimental measurements of diffusion coefficient profiles are
generally quite limited for most confined regions such as ion channels, nano-structures and nanopores. We
propose an alternative model to reduce number of Nernst-Planck equations to be
solved in complex chemical and
biological systems with multiple ion species by substituting Nernst-Planck
equations with Boltzmann distributions of ion concentrations. As such, we solve
the coupled Poisson-Boltzmann and Nernst-Planck (PBNP) equations, instead of
the PNP equations. The proposed PBNP equations are derived from a total energy
functional by using the variational principle. We
design a number of computational techniques, including the Dirichlet
to Neumann mapping, the matched interface and boundary (MIB), and relaxation
based iterative procedure, to ensure efficient solution of the proposed PBNP
equations. Two protein molecules, cytochrome c551 and Gramicidin A, are
employed to validate the proposed model under a wide range of bulk ion
concentrations and external voltages. Extensive numerical experiments show that
there is an excellent consistence between the results predicted from the
present PBNP model and those obtained from the PNP model in terms of the
electrostatic potentials, ion concentration profiles, and current-voltage (I-V)
curves. The present PBNP model is further validated by a comparison with
experimental measurements of I-V curves under various ion bulk concentrations. Numerical
experiments indicate that the proposed PBNP model is more efficient than the
original PNP model in terms of simulation time.
This work is published in Ref. [8].

Figure 5. Ion channel model [8,10]. Upper left: Gramicidin
A channel; Upper right: Electrostatic surface potential of the Gramicidin A
protein; Bottom left: The projection of the electrostatic potential along the
channel axis. Bottom right: Voltage-Current relations compared with
experimental data for the Gramicidin A channel.
[Application to proton
transport in ion channels]
Proton transport plays an important role in biological
energy transduction and sensory systems. Therefore it has attracted much
attention in biological science and biomedical engineering in the past few
decades. The present work proposes a multiscale/multiphysics model for
the understanding of the molecular mechanism of proton transport in transmembrane proteins involving continuum, atomic and
quantum descriptions, assisted with the generation and visualization
of membrane channel surfaces. We describe proton dynamics quantum mechanically
via a new density functional theory based on the Boltzmann statistics, while implicitly
model numerous solvent molecules as a dielectric continuum to reduce the number
of degrees of freedom. The density of all other ions in the solvent is assumed
to obey the Boltzmann distribution. The impact of protein molecular structure
and its charge polarization on the proton transport is considered explicitly at
the atomic scale. A variational solute-solvent
interface is designed to separate the explicit molecule and implicit solvent
regions. We formulate a total free energy functional to put proton kinetic and
potential energies, free energy of all other ions, polar and nonpolar energies
of the whole system on an equal footing. The variational
principle is employed to derive coupled governing equations for the proton
transport system. Generalized Laplace-Beltrami equation, generalized
Poisson-Boltzmann equation and generalized Kohn-Sham equation are obtained from
the present variational framework. The variational solvent-solute interface is generated and visualized
to facilitate the multiscale
discrete/continuum/quantum descriptions. Theoretical formulations for the
proton density and conductance are constructed based on fundamental laws of
physics. A number of mathematical algorithms, including the Dirichlet
to Neumann mapping (DNM), matched interface and boundary (MIB) method, Gummel iteration, and Krylov space
techniques are utilized to implement the
proposed model in a computationally efficient manner. The Gramicidin A (GA) channel is used to validate the performance of the
proposed proton transport model and demonstrate the efficiency of the proposed
mathematical algorithms. The proton channel conductance is studied over a
number of applied voltages and reference concentrations. A comparison with experimental
data verifies the present model predictions and confirms the proposed model.
This modeling and computation of proton transport are developed in Refs [12, 13,20] .
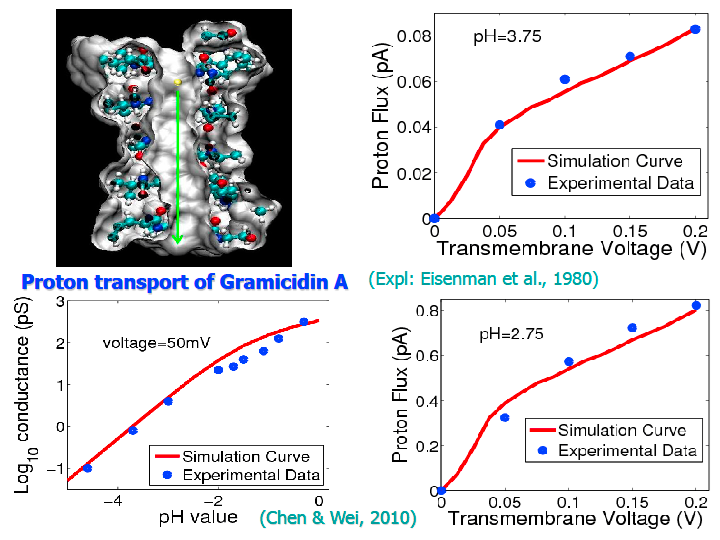
Figure 6. Simulation and comparison with
experimental data for proton transport in the Gramicidin A channel [12,13,20].
[Application to electron
transport in nano-devices]
The miniaturization of nano-scale
electronic devices, such as metal oxide semiconductor field effect transistors
(MOSFETs), has given rise to a pressing demand in the new theoretical
understanding and practical tactic for dealing with quantum mechanical effects
in integrated circuits. Modeling and simulation of this class of problems have
emerged as an important topic in applied and computational mathematics. Based
on the mathematical framework developed in our solvation analysis, we presents mathematical models and computational algorithms
for the simulation of nano-scale MOSFETs. We
introduce a unified two-scale energy functional to
describe the electrons and the continuum electrostatic potential of the nano-electronic device. This framework enables us to put
microscopic and macroscopic descriptions in an equal footing at nano scale. By optimization of the energy functional, we
derive consistently-coupled Poisson-Kohn-Sham equations. Additionally, layered
structures are crucial to the electrostatic and transport properties of nano transistors. A material interface model is proposed
for more accurate description of the electrostatics governed by the Poisson
equation. Finally, a new individual dopant model that utilizes the Dirac delta
function is proposed to understand the random doping effect in nano electronic devices. Two mathematical algorithms, the
matched interface and boundary (MIB) method and the Dirichlet-to-Neumann
mapping (DNM) technique, are introduced to improve the computational efficiency
of nano-device simulations. Electronic structures are
computed via subband decomposition and the transport
properties, such as the I-V curves and electron density, are evaluated via the
non-equilibrium Green's functions (NEGF) formalism. Two distinct device
configurations, a double-gate MOSFET and a four-gate MOSFET,
are considered in our three-dimensional numerical simulations. For these
devices, the current fluctuation and voltage threshold lowering effect induced
by the discrete dopant model are explored. Numerical convergence and model
well-posedness are also investigated in the present
work. This work is published in Ref. [6]
[Application to fast molecular
surface generation]
We
introduce a new framework for the surface generation based on the partial
differential equation (PDE) transform, which utilizes high order geometric
evolution equations. The PDE transform has recently been introduced by us as a
general approach for the mode decomposition of images, signals, and data. It
relies on the use of arbitrarily high-order PDEs to achieve the time–frequency
localization, control the spectral distribution, and regulate the spatial
resolution. The present work provides a new variational
derivation of high-order PDE transforms. The fast Fourier transform is utilized
to accomplish the PDE transform so as to avoid stringent stability constraints
in solving high-order PDEs. As a consequence, the time integration of high-order
PDEs can be done efficiently with the fast Fourier transform. The present
approach is validated with a variety of test examples in two-dimensional and
three-dimensional settings. We explore the impact of the PDE transform
parameters, such as the PDE order and propagation time, on the quality of
resulting surfaces. Additionally, we utilize a set of 10 proteins to compare
the computational efficiency of the present surface generation method and a
standard approach in Cartesian meshes. Moreover, we analyze the present method
by examining some benchmark indicators of biomolecular
surface, that is, surface area, surface-enclosed volume, solvation free energy,
and surface electrostatic potential. A test set of 13 protein molecules is used
in the present investigation. The electrostatic analysis is carried out via the
Poisson–Boltzmann equation model. To further demonstrate the utility of the
present PDE transform-based surface method, we solve the Poisson–Nernst–Planck
equations with a PDE transform surface of a protein. Second-order convergence
is observed for the electrostatic potential and concentrations. Finally, to
test the capability and efficiency of the present PDE transform-based surface
generation method, we apply it to the construction of an excessively large
biomolecule, a virus surface capsid. Virus surface morphologies of different
resolutions are attained by adjusting the propagation time. Therefore, the
present PDE transform provides a multiresolution
analysis in the surface visualization. Extensive numerical experiment and
comparison with an established surface model indicate that the present PDE
transform is a robust, stable, and efficient approach for biomolecular
surface generation in Cartesian. This work is described in Ref. [19]
[Application to mode
decomposition]
The
geometry flow formalism proposed in the present work is utilized to develop a
new method for the mode decomposition [15,16,17]. This
new method is called partial differential equation (PDE) transform [15]. Like
the wavelet transform, the PDE transform is able to decompose signals, images
and data into functional modes, such as edges, trend, texture and feature with
controllable frequency ranges and time-frequency localizations, which
correspond to appropriate multiresolution analysis in
the physical domain. Similarly, the PDE transform also has a prefect
reconstruction of original signals, images and data. The PDE transform has found its success in
signal processing [16,17],
biomedical image analysis [15] and biomolecular surface
construction and visualization [19].
[Computational algorithms]
Computational methods and algorithms
are designed and developed for the solution of the generalized
Poisson-Boltzmann equation and generalized geometric flow equations [1,2]. Specifically, a second-order finite difference scheme
is designed to solve the generalized Poisson-Boltzmann equation. The effect of
the appropriate preconditioner to the basic
Poisson-Boltzmann solver is explored. Both a simpleminded Euler method and an
appropriate alternative direction implicit (ADI) scheme are constructed to
solve the nonlinear generalized geometric flow equation. The ADI scheme allows
a relatively large time stepping and provides better efficiency. Finally, two
iterative approaches are designed and tested for the solution of the coupled
Poisson-Boltzmann equation and the generalized geometric flow equation. All
computational methods have been extensively validated [1,2].
Second order convergent numerical
method has been developed to solve the Poisson-Nernst-Planck (PNP) equations
against complex protein surfaces, geometric singularities, and singular delta
functions [10]. This is the first second order solver for the PNP equations in biomolecular context.
The matched interface and boundary (MIB)
developed in Wei’s lab has been further studied to account for solving the
Poisson-Boltzmann equation with multiple material interfaces, which occur in
implicit solvation models when metal active centers exist. This is the first
time the second order convergent numerical method has been constructed for this
class of problems. This work is described in Refs. [9,14].
We have been putting effort on the development of
efficient computational methods and software packages for the biophysical and
biochemistry communities. The MIBPB package described in our recent paper [5]
has been further improved by considering a full scope of topological variations
in complex biomolecules. Previously, geometric singularities of large proteins may
cause a stop of our algorithm, although on a rare basis. We have fixed the algorithm problem and
developed a robust interface technique based Poisson-Boltzmann solver
package.
We are developing a finite element version of our
MIB method and comparing this approach with our earlier finite difference based
MIB. A three-dimensional MIB Galerkin method is under
construction. Part of our work is described in Refs. [24,25].
[Software development]
We have developed a software package
for the solution of the Poisson-Boltzmann equation [5]. The Poisson-Boltzmann equation (PBE) is an
established model for the electrostatic solvation analysis of biomolecules. The
development of advanced computational techniques for the solution of the PBE
has been an important topic in the past two decades. This work presents a
matched interface and boundary (MIB) based PBE software package, the MIBPB
solver, for electrostatic analysis. The MIBPB has a unique feature that it is
the first interface technique based PBE solver that rigorously enforces the
solution and flux continuity conditions at the dielectric interface between the
biomolecule and the solvent. For protein molecular surfaces which may possess
troublesome geometrical singularities, the MIB scheme makes the MIBPB by far
the only existing PBE solver that is able to deliver the second order
convergence, i.e., the accuracy increases four times when the mesh size is
halved. The MIBPB method is also equipped with a Dirichlet-to-Neumann
mapping (DNM) technique, that builds a Green's function approach to
analytically resolve the singular charge distribution in biomolecules in order
to obtain reliable solutions at meshes as coarse as 1![]() while it usually takes other traditional PB
solvers 0.25
while it usually takes other traditional PB
solvers 0.25![]() to reach similar level of reliability.
The present work further accelerates the rate of convergence of linear equation
systems resulting from the MIBPB by utilizing the Krylov
subspace (KS) techniques. Condition numbers of the MIBPB matrices are signi_cantly reduced by using appropriate Krylov subspace solver and preconditioner
combinations. Both linear and nonlinear PBE solvers in the MIBPB package are
tested by protein-solvent solvation energy calculations and analysis of salt
effects on protein-protein binding energies, respectively.
to reach similar level of reliability.
The present work further accelerates the rate of convergence of linear equation
systems resulting from the MIBPB by utilizing the Krylov
subspace (KS) techniques. Condition numbers of the MIBPB matrices are signi_cantly reduced by using appropriate Krylov subspace solver and preconditioner
combinations. Both linear and nonlinear PBE solvers in the MIBPB package are
tested by protein-solvent solvation energy calculations and analysis of salt
effects on protein-protein binding energies, respectively.
[Report from Nathan
Baker group]
- We received the FORTRAN
code from Zhan and Guowei Wei in January 2011
and slightly modified it to implement appropriate interaction potentials
and non-polar free energy.
- In a paper recently submitted to Journal of Computational Chemistry
[26], we have developed a new parameterization scheme for our geometric
flow approach. Implicit solvent models are popular for their high
computational efficiency and simplicity over explicit solvent models and
are extensively used for computing molecular solvation properties.
The accuracy of implicit solvent models depends on the geometric
description of the solute-solvent interface and the solvent dielectric
profile that is defined near the surface of the solute molecule.
Typically, it is assumed that the dielectric profile is spatially
homogeneous in the bulk solvent medium and varies sharply across the
solute-solvent interface. However, the specific form of this profile
is often described by ad hoc geometric models rather than physical
solute-solvent interactions. Hence, it is of significant interest to
improve the accuracy of these implicit solvent models by more
realistically defining the solute-solvent boundary within a continuum
setting. Recently, a differential geometry-based geometric flow
solvation model was developed, in which the polar and nonpolar free
energies are coupled through a characteristic function that describes a
smooth dielectric interface profile across the solvent–solute boundary in
a thermodynamically self-consistent fashion. The main parameters of
the model are the solute/solvent dielectric constants, solvent pressure on
the solute, microscopic surface tension, solvent density, and molecular
force-field parameters. In this work, we investigate how changes in
the pressure, surface tension, and choice of different force-field charge
and radii parameters affect the prediction accuracy for hydration free
energies of 17 small organic molecules based on the geometric flow
solvation model. These results are highlighted in the following
Figure 7 and provide insights on the parameterization, accuracy, and
predictive power of this new implicit solvent model.
![Description: Figure-2[1]](research_files/image034.jpg)
Figure 7. Solvation errors for 17 small
molecules generated by new parameterizations of the differential geometry based
solvation model by Baker’s group.
C.
Future Plan
In our future plan, we will explore a number of
fronts. The first is to further advance the application of the present
geometric flow framework to geometric and topological modeling of biomolecular systems. Additionally, we will construct
differential geometry based models and methods for nanofluidic
systems. Finally, we will continue our computational algorithm and software
development.
First, geometric modeling and topological modeling of
macromolecular systems are important tasks in structural biology and molecular
biology. The differential geometry based methods and techniques developed in
the present work can play a crucial role in such modeling. The essential idea is to combine geometric
flow evolution equations to improve the quality of biomolecular
data collected by the Cryo Electron Microscopy, which
is of low resolution and often corrupted with noise. The experience learned
from the present project will enable us to do an excellent job in geometric and
topological modeling of macromolecules, including improved meshing, a better
estimation of area and volume, robust calculation of curvature, genus number,
and Frenet frame.
Additionally, nano-fluidics
is a fast-growing nano-bio technology. It has been
applied to a wide range of chemical and biological systems, including DNA
sequencing, PCR, separation of protein-antibody, membrane protein
crystallization, etc. Most current theories describe only the flow effect, but
ignore the electrostatic interactions. A multiscale
model has been proposed in our recent work [3]. One of our future interests is
to carry detail numerical analysis and simulation of nanofluidic
systems by using the proposed differential geometry based multiscale
models.
The Baker group will continue making contribution to solvation
modeling and software development. The newly developed differential geometry
based solvation codes will be integrated with the popular software tools
developed in the Baker group, such as APBS and PDB2PQR. This development will
ensure that the results obtained from this NIH grant will be immediately
available to the computational biology community.
Peter Bates will continue his effort in the mathematical analysis of the proposed
coupled Poisson-Boltzmann and geometric flow equations. Mathematical analysis
usually follows a different path. Its progress is normally slow, but runs
deep. We expect that our future
mathematical analysis will lead to better understanding of theoretical and
computational aspects of the proposed multiscale solvation
models.
D. Publication
- Zhan Chen, Nathan Baker and G.
W. Wei, Differential geometry
based solvation model I: Eulerian formulation,
Journal of Computational Physics, 229,
8231-8258 (2010). NIHMSID#215329
- Zhan Chen, Nathan Baker and G.
W. Wei, Differential geometry
based solvation model II: Lagrangian
formulation, Journal of Mathematical biology, in press (2011). NIHMSID#215331
- Guo-Wei,
Wei, Differential geometry based multiscale models, Bulletin of Mathematical Biology,
volume 72, 1562-1622, (2010). NIHMSID#
215327 http://www.springerlink.com/content/8303641145x84470/fulltext.pdf
- Changjun
Chen, Rishu Saxena, and Guo-Wei Wei, Differential
geometry based multiscale model for virus
capsid dynamics, Int.
J. Biomed Imaging, Volume
2010, Article ID 308627, 308627, 9 pages (2010). (NIHMSID#215328
http://www.hindawi.com/journals/ijbi/2010/308627.html
- Duan
Chen, Zhan Chen, Changjun Chen, Weihua Geng and
Guo-Wei Wei, MIBPB: A software package for
electrostatic analysis, Journal of computational Chemistry, 32,
756–770 (2011). NIHMSID#215330
- Duan Chen and Guo-Wei
Wei, Modeling and
simulation of nano-electronic
devices, Journal of Computational Physics,
229, 4431-4460, (2010). NIHMSID#181011
- Weihua Geng and G.W. Wei, Multiscale molecular dynamics
via the matched
interface and boundary
(MIB) method, Journal of Computational Physics,
230, 435-457 (2011). NIHMSID#243881
- Qiong Zheng and Guo-Wei Wei,
Poisson-Boltzmann-Nernst-Planck
model. Journal of Chemical Physics, 134 (19), 194101, (2011). NIHMSID#298927
- Kelin
Xia, Meng
Zhan and Guo-Wei Wei,
The matched interface and
boundary (MIB) method for
multi-domain elliptic
interface problems. Journal of Computational Physics , 230, 8231–8258 (2011). NIHMSID#281679
- Qiong Zheng, Duan Chen and Guo-Wei Wei,
Second-order
Poisson-Nernst-Planck solver
for ion transport. Journal of Computational Physics, 230, 5239-5262 (2011). NIHMSID#286113
- Zhan Chen and G. W. Wei, Differential geometry
based salvation model
III: Quantum formulation, Journal of Chemical
Physics, 135, 194108 (2011). NIHMSID#374656
- Duan
Chen, Zhan Chen
and Guo-Wei Wei,
Quantum dynamics in
continuum models for
proton transport II:
Variational solvent-solute interface, International Journal
for Numerical
Methods in Biomedical
Engineering, 28, 25-51 (2012). PMCID: PMC3274368
- Duan Chen
and Guo-Wei Wei, Quantum dynamics in continuum models
for proton
transport I: Basic
formulation, Communication in
Computational
Physics, in press
(2012). NIHMSID
#374856
- Kelin Xia, Meng
Zhan, Decheng Wan and
Guo-Wei
Wei, Adaptively deformed
mesh based interface method for elliptic equations with discontinuous
coefficients, Journal of
Computational Physics, 231, 1440–1461 (2012). NIHMSID# 374658
- Yang Wang, Guo-Wei Wei
and Siyang Yang, Partial differential equation
transform --- variational formulation and
Fourier analysis , International
Journal for Numerical Methods in Biomedical
Engineering, 27, 1996-2020, (2011). NIHMSID#298929
- Yang Wang, Guo-Wei Wei
and Siyang Yang,
Model decomposing evolution equations, Journal of Scientific Computing, 50,
495-518 (2012). PMCID: PMC3293488
- Yang Wang,
Guo-Wei
Wei and Siyang Yang, Iterative filtering decomposition
using local spectral
evolution kernels, Journal of Scientific Computing, 50, 629-664 (2012), PMCID: PMC3281768
- Yang Wang, Guo-Wei
Wei and Siyang Yang, Selective extraction
of entangled textures
via adaptive PDE transform, Int.
J. Biomed Imaging, Volume
2012 (2012), Article ID 958142(2012). PMCID: PMC3272340
- Qiong
Zheng, Siyang Yang and Guo-Wei Wei, Molecular surface
generation using
PDE
transform. International Journal for
Numerical Methods in Biomedical Engineering, 28, 291-316
(2012). NIHMSID#329786
- Duan Chen and Guo-Wei Wei, Quantum dynamics in continuum models for proton transport --- Generalized
correlation, Journal of Chemical Physics, 136, 134109 (2012) NIHMSID#256945
- G.W. Wei, Zhan Chen, Kelin Xia and
Qiong Zheng, Variational multiscale models
for charge transport, SIAM review. In press (2012). NIHMSID#367755
- Zhan Chen, Shan Zhao,
Jaehun Chun, Dennis G. Thomas, Nathan
Baker, Peter Bates and Guo-Wei Wei, Variational
multiscale approach
for
nonpolar solvation analysis, (2012). NIHMSID#374673
- Langhua Hu and Guo-Wei
Wei, Nonlinear Poisson
equation for inhomogeneous media. (2012).
- Kelin
Xia, Meng Zhan, and Guo-Wei
Wei, MIB Galerkin for
elliptic interface problems
(2012).
- L. Mu,
J. P. Wang, G. W. Wei,
X. Ye and S.Zhao, Weak
Galerkin methods for
second order elliptic
interface problems. (2012)
- Dennis
G. Thomas, Jaehun Chun, Zhan Chen, Guowei Wei, Nathan A. Baker, Parameterization of a
Geometric Flow Implicit Solvation Model, submitted to Journal of Computational
Chemistry. (2012)